1月11日訊 過(guò)去的2020年,是我國(guó)全面建成小康社會(huì)和“十三五”規(guī)劃收官之年,中國(guó)醫(yī)改繼續(xù)向深水區(qū)挺進(jìn),深化三醫(yī)聯(lián)動(dòng)改革。2020年也是非同尋常的一年,國(guó)家積極應(yīng)對(duì)新冠疫情,上半年發(fā)布了大量疫情政策。7月,國(guó)務(wù)院發(fā)布《關(guān)于印發(fā)深化醫(yī)藥衛(wèi)生體制改革2020年下半年重點(diǎn)工作任務(wù)的通知》,下半年醫(yī)改工作緊鑼密鼓展開(kāi),配套政策相應(yīng)發(fā)布。以上兩方面的因素使2020年成為發(fā)布醫(yī)改政策數(shù)量最多的一年。
本年度國(guó)家層面發(fā)布醫(yī)藥行業(yè)相關(guān)政策超600條,其中醫(yī)藥相關(guān)政策約占總數(shù)的一半,藥監(jiān)局作為醫(yī)藥政策發(fā)布的主要部門(mén),在各發(fā)文機(jī)構(gòu)中政策發(fā)布數(shù)量居首。面對(duì)浩繁的政策文件,囿于篇幅,本文聚焦于醫(yī)藥政策中與藥品審評(píng)審批相關(guān)的主要政策,尤其是加速創(chuàng)新藥審評(píng)審批的配套細(xì)則。
一、綱領(lǐng)性法規(guī)公布
2020年3月30日,國(guó)家市場(chǎng)監(jiān)督管理總局公布了2020新版《藥品注冊(cè)管理辦法》和《藥品生產(chǎn)監(jiān)督管理辦法》,其中注冊(cè)管理辦法是在2007版基礎(chǔ)上,時(shí)隔13年之后的重大修訂。本次修改內(nèi)容涉及藥品注冊(cè)分類(lèi)改革、優(yōu)化審評(píng)審批流程、強(qiáng)化責(zé)任追究等方面。兩份綱領(lǐng)性文件已于2020年7月1日起正式施行?!端幤纷?cè)管理辦法》(下稱(chēng)“辦法”)的要點(diǎn)包括以下五個(gè)方面。
優(yōu)化和提高審評(píng)效率
辦法強(qiáng)調(diào)以臨床價(jià)值為導(dǎo)向,鼓勵(lì)研究和創(chuàng)新制藥,積極推動(dòng)仿制藥發(fā)展。國(guó)家藥監(jiān)局持續(xù)推進(jìn)審評(píng)審批制度改革,優(yōu)化程序,提高效率,建立以審評(píng)為主導(dǎo),檢驗(yàn)、核查、監(jiān)測(cè)與評(píng)價(jià)等為支撐的藥品注冊(cè)管理體系。
四大通道加快新藥上市
對(duì)標(biāo)美國(guó)FDA,國(guó)家藥監(jiān)局建立了四條特殊審評(píng)通道:突破性治療藥物、附條件批準(zhǔn)、優(yōu)先審評(píng)審批、特別審批程序。定位于藥品臨床研發(fā)階段的是突破性治療藥物程序;定位于藥品上市注冊(cè)階段的是附條件批準(zhǔn)和優(yōu)先審評(píng)審批程序;特別審批程序針對(duì)突發(fā)公共衛(wèi)生事件。
進(jìn)一步縮短審評(píng)時(shí)間
藥物臨床試驗(yàn)申請(qǐng)改為自受理之日起60日內(nèi)決定是否同意,生物等效性試驗(yàn)備案即可實(shí)施。藥品上市許可申請(qǐng)審評(píng)時(shí)限為200日,其中優(yōu)先審評(píng)審批為130日,臨床急需境外已上市罕見(jiàn)病用藥為70日;單獨(dú)申報(bào)仿制境內(nèi)已上市化學(xué)原料藥為200日;藥品再注冊(cè)審查為120日。
全面落實(shí)藥品上市許可持有人制度
明確上市許可持有人(MAH)為能夠承擔(dān)相應(yīng)責(zé)任的企業(yè)或藥品研制機(jī)構(gòu)等,要求建立藥品質(zhì)量保證體系,對(duì)藥品的全生命周期進(jìn)行管理,開(kāi)展上市后研究,承擔(dān)上市藥品的安全有效和質(zhì)量責(zé)任。
強(qiáng)調(diào)原輔包關(guān)聯(lián)審批
在審批藥品制劑時(shí),對(duì)化學(xué)原料藥、輔料及直接接觸藥品的包裝材料和容器進(jìn)行關(guān)聯(lián)審評(píng)。仿制境內(nèi)已上市藥品所用的化學(xué)原料藥的,可以申請(qǐng)單獨(dú)審評(píng)審批。
為了保障2020版《藥品注冊(cè)管理辦法》順利施行,國(guó)家藥監(jiān)局相繼出臺(tái)了系列藥品注冊(cè)分類(lèi)、藥品審評(píng)等配套文件的征求意見(jiàn)稿,加快藥品審評(píng)審批工作,提高藥物可及性和企業(yè)研發(fā)新藥的積極性。
二、2020版化藥、生物制品、中藥注冊(cè)分類(lèi)出臺(tái)
2020年6月30日,國(guó)家藥監(jiān)局發(fā)布化學(xué)藥品、生物制品注冊(cè)分類(lèi)及申報(bào)資料要求的通告,注冊(cè)分類(lèi)自2020年7月1日起實(shí)施,申報(bào)資料要求自2020年10月1日起實(shí)施。2020年9月,國(guó)家藥監(jiān)局發(fā)布了中藥注冊(cè)分類(lèi)及申報(bào)資料要求的通告,自2021年1月1日起,一律按新要求提交申報(bào)資料。
圖表1. 2020版藥品注冊(cè)分類(lèi)基本框架
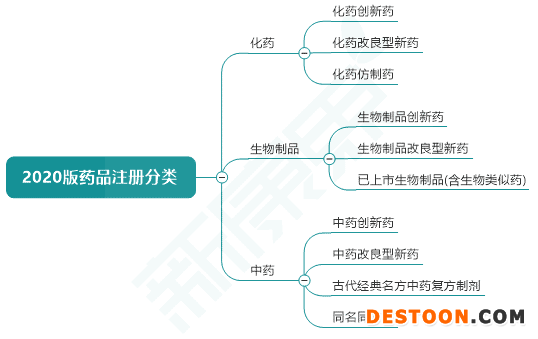
來(lái)源:國(guó)家藥監(jiān)局,中康產(chǎn)業(yè)資本研究中心
2020版化藥注冊(cè)分類(lèi)
該版本與2016版化藥注冊(cè)分類(lèi)差別不大,主要改動(dòng)有3點(diǎn):“原料藥及制劑”、“制劑”這些詞語(yǔ)統(tǒng)一為“藥品”;2類(lèi)、3類(lèi)和4類(lèi)藥品的定義中強(qiáng)調(diào)了一致性評(píng)價(jià);5.1分類(lèi)中增加了改良型新藥和境內(nèi)上市的境外原研藥品新增適應(yīng)癥在國(guó)內(nèi)申報(bào)上市。
2020版化藥注冊(cè)分類(lèi)分為5類(lèi),基本內(nèi)容為:1類(lèi)為境內(nèi)外均未上市的創(chuàng)新藥;2類(lèi)為境內(nèi)外均未上市的改良型新藥;3類(lèi)為仿制境外上市但境內(nèi)未上市原研藥品的藥品;4類(lèi)為仿制已在境內(nèi)上市原研藥品的藥品;5類(lèi)為境外上市的藥品申請(qǐng)?jiān)诰硟?nèi)上市。
2020版生物制品注冊(cè)分類(lèi)
預(yù)防用生物制品、治療用生物制品,以及按生物制品管理的體外診斷試劑的分類(lèi)均依據(jù)創(chuàng)新程度和境內(nèi)外上市的情況分類(lèi)。預(yù)防用生物制品分為創(chuàng)新型疫苗(1類(lèi))、改良型疫苗(2類(lèi))、境內(nèi)或境外已上市的疫苗(3類(lèi)),每個(gè)分類(lèi)下還有子類(lèi)。類(lèi)似地,治療用生物制品也分為3類(lèi)。
藥品注冊(cè)分類(lèi)是給藥品定位的重要指標(biāo),對(duì)藥品臨床試驗(yàn)、申報(bào)、定價(jià)、準(zhǔn)入等方面都有影響。在鼓勵(lì)創(chuàng)新的背景下,1類(lèi)新藥可以納入優(yōu)先審評(píng)審批程序,上市后市場(chǎng)準(zhǔn)入通道順暢,且具有相對(duì)較高的定價(jià)。4類(lèi)仿制藥則基本沒(méi)有優(yōu)待,而且需要經(jīng)歷帶量采購(gòu)的洗禮,大幅降價(jià)后幾乎只能獲取生產(chǎn)利潤(rùn)。新版藥品注冊(cè)分類(lèi)更加嚴(yán)格地依據(jù)創(chuàng)新程度,聯(lián)合新版《藥品注冊(cè)管理辦法》和《藥品生產(chǎn)監(jiān)督管理辦法》,使不同創(chuàng)新程度的藥品得到適當(dāng)?shù)墓芾怼?/div>




2020版中藥注冊(cè)分類(lèi)
在中藥注冊(cè)分為4類(lèi):中藥創(chuàng)新藥、中藥改良型新藥、古代經(jīng)典名方中藥復(fù)方制劑、同名同方藥,其中前三類(lèi)屬于中藥新藥。注意中藥分類(lèi)不代表藥物研制水平及療效的高低,僅表明注冊(cè)申報(bào)資料要求不同。為了加強(qiáng)對(duì)古典醫(yī)籍精華的梳理和挖掘,3類(lèi)中藥細(xì)分為按古代經(jīng)典名方目錄管理的中藥復(fù)方制劑(3.1類(lèi))及其他來(lái)源于古代經(jīng)典名方的中藥復(fù)方制劑(3.2類(lèi)),其中3.2類(lèi)包括未按古代經(jīng)典名方目錄管理的古代經(jīng)典名方中藥復(fù)方制劑和基于古代經(jīng)典名方加減化裁的中藥復(fù)方制劑。這也體現(xiàn)了國(guó)家對(duì)中藥行業(yè)的支持與鼓勵(lì)。
三、中國(guó)藥品專(zhuān)利糾紛早期解決機(jī)制首次出臺(tái)
2020年9月11日,國(guó)家藥監(jiān)局、國(guó)家知識(shí)產(chǎn)權(quán)局發(fā)布了《藥品專(zhuān)利糾紛早期解決機(jī)制實(shí)施辦法(試行)(征求意見(jiàn)稿)》(下稱(chēng)“意見(jiàn)稿”),進(jìn)一步完善專(zhuān)利鏈接制度。該政策的目的是盡量在專(zhuān)利訴訟前解決藥品專(zhuān)利糾紛,以減少資源浪費(fèi),加快仿制藥對(duì)專(zhuān)利藥的合理替代。在保護(hù)藥品專(zhuān)利權(quán)人的合法權(quán)益的前提下,促進(jìn)高水平仿制藥(首仿藥)研發(fā),同時(shí)刺激研發(fā)更具原創(chuàng)性的新藥。
2017年10月,中辦、國(guó)辦印發(fā)了中國(guó)藥監(jiān)政策改革的綱領(lǐng)性文件之一《關(guān)于深化審評(píng)審批制度改革鼓勵(lì)藥品醫(yī)療器械創(chuàng)新的意見(jiàn)》,即赫赫有名的42號(hào)文,其中就要求探索建立藥品專(zhuān)利鏈接制度。隨后幾年內(nèi),多個(gè)機(jī)構(gòu)多次發(fā)文推進(jìn)。本次發(fā)布的意見(jiàn)稿是對(duì)前期政策的延續(xù),也是我國(guó)首次出臺(tái)藥品專(zhuān)利糾紛早期解決機(jī)制。
為了推動(dòng)我國(guó)仿制藥高質(zhì)量發(fā)展,意見(jiàn)稿從多個(gè)方面探索藥品專(zhuān)利糾紛早期解決辦法,包括建立中國(guó)上市藥品專(zhuān)利信息等級(jí)平臺(tái)、明確藥品專(zhuān)利信息等級(jí)范圍、規(guī)定仿制藥申請(qǐng)人專(zhuān)利狀態(tài)聲明制度、明確專(zhuān)利人或利害人提出異議的實(shí)現(xiàn)、對(duì)化學(xué)藥品設(shè)置審評(píng)審批等待期、對(duì)藥品審評(píng)審批實(shí)施分類(lèi)處理,以及給予專(zhuān)利挑戰(zhàn)成功的首仿藥市場(chǎng)獨(dú)占期等。
值得著重指出的是,意見(jiàn)稿第11條規(guī)定:“對(duì)首個(gè)挑戰(zhàn)專(zhuān)利成功且首個(gè)獲批上市的化學(xué)仿制藥品,給予市場(chǎng)獨(dú)占期,國(guó)務(wù)院藥品監(jiān)督管理部門(mén)在該藥品獲批之日起12個(gè)月內(nèi)不再批準(zhǔn)同種仿制藥上市,市場(chǎng)獨(dú)占期不超過(guò)被挑戰(zhàn)藥品的專(zhuān)利權(quán)期限。市場(chǎng)獨(dú)占期內(nèi)國(guó)藥藥品審評(píng)機(jī)構(gòu)不停止技術(shù)審評(píng)。對(duì)技術(shù)申請(qǐng)通過(guò)的化學(xué)仿制藥注冊(cè)申請(qǐng),待市場(chǎng)獨(dú)占期到期前20個(gè)工作日將相關(guān)化學(xué)仿制藥申請(qǐng)轉(zhuǎn)入行政審批環(huán)節(jié)”。意見(jiàn)稿基本參照了美國(guó)FDA的一系列做法,但是對(duì)仿制藥專(zhuān)利挑戰(zhàn)的鼓勵(lì)力度更大,最大12個(gè)月的市場(chǎng)獨(dú)占期是美國(guó)專(zhuān)利挑戰(zhàn)成功的首仿藥6個(gè)月市場(chǎng)獨(dú)占期的2倍。這將極大調(diào)動(dòng)藥企的研發(fā)熱情,以專(zhuān)利挑戰(zhàn)模式布局首仿,有助于提升仿制藥研發(fā)質(zhì)量,同時(shí)以低于被挑戰(zhàn)原研藥的價(jià)格普惠患者。
四、新藥審評(píng)審批的加速通道政策細(xì)則落地
2020年7月8日,國(guó)家藥監(jiān)局發(fā)布了《突破性治療藥物審評(píng)工作程序(試行)》、《藥品附條件批準(zhǔn)上市審評(píng)審批工作程序(試行)》和《藥品上市許可優(yōu)先審評(píng)審批工作程序(試行)》三項(xiàng)配套細(xì)則文件,以配合2020版《藥品注冊(cè)管理辦法》中加快新藥上市注冊(cè)程序的實(shí)施。
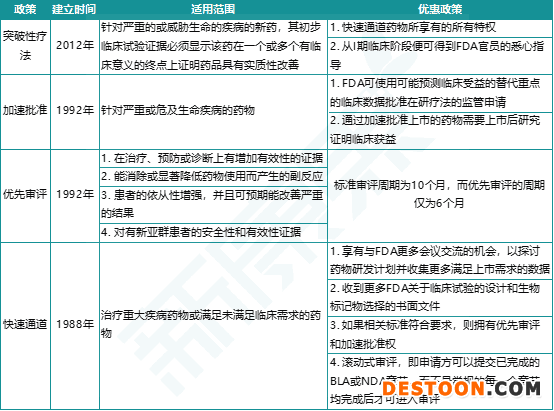
這3項(xiàng)工作程序是我國(guó)鼓勵(lì)新藥研發(fā)、加速新藥上市而設(shè)立的3個(gè)特殊審評(píng)通道,參照了美國(guó)FDA的4個(gè)特殊審評(píng)通道:突破性療法通道(breakthrough therapy)、加速批準(zhǔn)(accelerated approval)、優(yōu)先審評(píng)(priority review)和快速通道(fast track)。“突破性療法”要求在初步臨床試驗(yàn)中看到藥物在一個(gè)或多個(gè)有臨床意義的終點(diǎn)上較現(xiàn)有療法有顯著改善;“加速批準(zhǔn)”可依據(jù)臨床試驗(yàn)的替代終點(diǎn)或中間終點(diǎn)來(lái)批準(zhǔn)藥物;“優(yōu)先審評(píng)”將上市申請(qǐng)的審評(píng)周期從標(biāo)準(zhǔn)的10個(gè)月縮短至6個(gè)月;“快速通道”則允許藥企在研發(fā)過(guò)程中滾動(dòng)提交上市申請(qǐng)資料。
圖表2. FDA加快創(chuàng)新藥上市的4種特殊審評(píng)通道
來(lái)源:FDA,中康產(chǎn)業(yè)資本研究中心
FDA設(shè)置特殊審評(píng)通道的目的是促進(jìn)有重要治療價(jià)值的創(chuàng)新藥盡快上市,同時(shí)保證在審評(píng)過(guò)程中不會(huì)損害藥品的安全性和有效性標(biāo)準(zhǔn)。美國(guó)之所以成為全球每年獲批創(chuàng)新藥最多的國(guó)家,其中一個(gè)重要原因就是FDA在新藥審評(píng)上設(shè)置的加速通道。
雖然中國(guó)的藥監(jiān)機(jī)構(gòu)學(xué)習(xí)了FDA的加速通道模式,但是中國(guó)的幾種特殊審評(píng)通道政策的引入與發(fā)展與中國(guó)創(chuàng)新藥研發(fā)總體情況的發(fā)展密切相關(guān),體現(xiàn)了側(cè)重點(diǎn)從“量”到“質(zhì)”的轉(zhuǎn)變。
中國(guó)創(chuàng)新藥加速通道之一:優(yōu)先審評(píng)審批
優(yōu)先審評(píng)審批發(fā)端于中國(guó)藥監(jiān)政策改革的綱領(lǐng)文件之一,2015年8月發(fā)布的“44號(hào)文”《國(guó)務(wù)院關(guān)于改革藥品醫(yī)療器械審評(píng)審批制度的意見(jiàn)》,其中提出加快創(chuàng)新藥審評(píng)審批。2016年2月,原國(guó)家食藥監(jiān)總局(CFDA)發(fā)布關(guān)于優(yōu)先解決藥品注冊(cè)申請(qǐng)積壓實(shí)行優(yōu)先審評(píng)的意見(jiàn),覆蓋國(guó)內(nèi)外未上市的創(chuàng)新藥、臨床急需的高質(zhì)量層次仿制藥、對(duì)腫瘤、兒童用藥、罕見(jiàn)病、肺結(jié)核、病毒性肝炎等疾病領(lǐng)域創(chuàng)新藥。2020版《藥品上市許可優(yōu)先審評(píng)審批工作程序(試行)》則是基于2017年12月28日發(fā)布的《關(guān)于鼓勵(lì)藥品創(chuàng)新實(shí)行優(yōu)先審評(píng)審批的意見(jiàn)》。在2020版工作程序發(fā)布之前,政策傾向于解決有臨床價(jià)值的創(chuàng)新藥和臨床急需的高水平仿制藥在“量”上的問(wèn)題,配合解決藥品注冊(cè)申請(qǐng)積壓的問(wèn)題。在審評(píng)積壓?jiǎn)栴}基本解決,國(guó)內(nèi)臨床需求基本滿(mǎn)足之后,優(yōu)先審評(píng)審批程序適用范圍則更加傾向于具有明顯臨床價(jià)值的新藥。
圖表3. 2020年和2017年優(yōu)先審評(píng)審批程序適用范圍
來(lái)源:國(guó)家藥監(jiān)局,中康產(chǎn)業(yè)資本研究中心
中國(guó)創(chuàng)新藥加速通道之二:突破性治療藥物程序
中國(guó)藥監(jiān)局引入與FDA的“突破性療法通道”類(lèi)似的“突破性治療藥物程序”較晚,2019年11月藥審中心(CDE)發(fā)布了《突破性治療藥物工作程序(征求意見(jiàn)稿)》。2020版《藥品注冊(cè)管理辦法》中納入了突破性治療藥物的相關(guān)規(guī)定,2020年7月發(fā)布的《突破性治療藥物審評(píng)工作程序(試行)》便是與之配套的細(xì)則。
可申請(qǐng)突破性治療藥物認(rèn)定的藥品需要同時(shí)滿(mǎn)足兩個(gè)條件:(1)用于防治嚴(yán)重危及生命或者嚴(yán)重影響生存質(zhì)量的疾病;(2)尚無(wú)有效防治手段或者與現(xiàn)有治療手段相比有足夠證據(jù)表明具有明顯臨床優(yōu)勢(shì)的創(chuàng)新藥或者改良型新藥等。藥審中心對(duì)納入突破性治療藥物程序的藥物優(yōu)先配置資源進(jìn)行溝通交流,加強(qiáng)指導(dǎo),且在上市申請(qǐng)階段納入優(yōu)先審評(píng)審批。優(yōu)厚的待遇意味著突破性治療藥物的篩選更加嚴(yán)格。
突破性治療藥物認(rèn)定要求初步臨床試驗(yàn)證明,藥物在一個(gè)或多個(gè)有臨床意義的指標(biāo)上,較現(xiàn)有療法有顯著改善。具有臨床意義的終點(diǎn)通常指與疾病發(fā)生、發(fā)展、死亡和功能相關(guān)的終點(diǎn),也可以包括經(jīng)過(guò)驗(yàn)證的替代終點(diǎn)、可能預(yù)測(cè)臨床獲益的替代終點(diǎn)或者中間臨床終點(diǎn)、安全性終點(diǎn)等。一般而言,替代終點(diǎn)、中間臨床終點(diǎn)、安全性終點(diǎn)的較常規(guī)臨床終點(diǎn)可更易收集、更快達(dá)到,因此有助于加快創(chuàng)新藥的研發(fā)進(jìn)程。需要注意的是,藥物納入突破性治療藥物程序并不意味著一定可以獲批上市,CDE如果發(fā)現(xiàn)納入程序的藥物臨床試驗(yàn)不再符合納入條件時(shí),將啟動(dòng)終止程序。
圖表4. 突破性治療藥物程序規(guī)定具有明顯臨床優(yōu)勢(shì)的情形
來(lái)源:CDE,中康產(chǎn)業(yè)資本研究中心
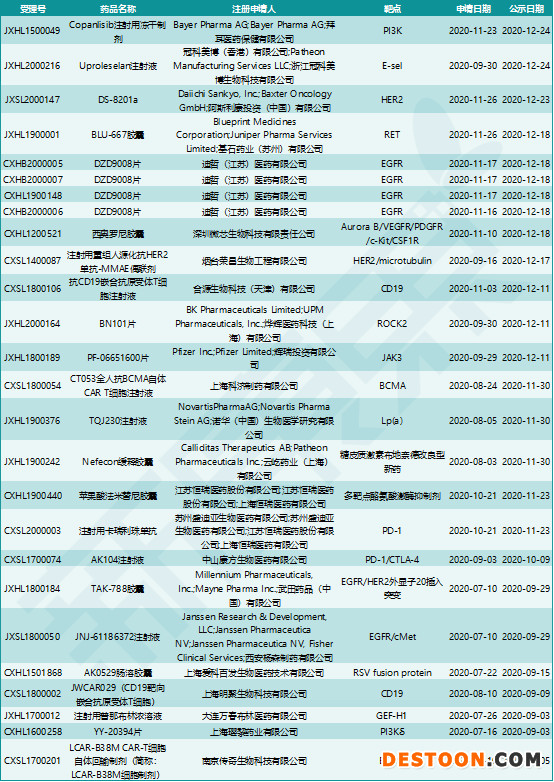
突破性治療藥物程序得到創(chuàng)新藥研發(fā)企業(yè)的熱烈響應(yīng)。在7月8日發(fā)布工作程序后,7月10日就有3家企業(yè)提交了申請(qǐng),其中傳奇生物靶向BCMA的CAR-T療法LCAR-B38M在8月12日取得了國(guó)家藥監(jiān)局首個(gè)突破性治療藥物認(rèn)定。
從7月10日至2020年底,在不到6個(gè)月的時(shí)間內(nèi),共有23款創(chuàng)新藥被CDE授予突破性治療藥物認(rèn)定。這些藥物類(lèi)型廣泛,有CAR-T療法、ADC、雙抗、ASO(反義單鏈寡核苷酸),小分子藥物靶點(diǎn)包括RET、EGFR、JAK、ROCK2、PI3Kδ等。這些創(chuàng)新藥的研發(fā)企業(yè)既有跨國(guó)藥企如輝瑞、諾華、武田等,也有本土藥企如恒瑞醫(yī)藥、康方生物、科技生物等。
圖表5. 2020年納入突破性治療藥物程序的創(chuàng)新藥
來(lái)源:CDE,中康產(chǎn)業(yè)資本研究中心
中國(guó)創(chuàng)新藥加速通道之三:附條件批準(zhǔn)上市
2007版《藥品注冊(cè)管理辦法》就提到對(duì)艾滋病、惡性腫瘤、罕見(jiàn)病等疾病且具有明顯臨床治療優(yōu)勢(shì)的新藥實(shí)行特殊審批,這是我國(guó)關(guān)于附條件批準(zhǔn)的最早政策表述。在后續(xù)的十多年間,僅有少數(shù)藥品被納入附條件批準(zhǔn)通道。2019年11月,CDE發(fā)布了《臨床急需藥品附條件批準(zhǔn)上市技術(shù)指導(dǎo)原則(征求意見(jiàn)稿)》,其中對(duì)藥企如何使用附條件批準(zhǔn)通道給出了詳細(xì)的指導(dǎo)。2020版《藥品注冊(cè)管理辦法》正式納入了附條件申請(qǐng)的政策,同時(shí)規(guī)定納入附條件批準(zhǔn)通道的藥品可申請(qǐng)優(yōu)先審評(píng)審批。
我國(guó)的附條件批準(zhǔn)通道借鑒了FDA的加速批準(zhǔn)(accelerated approval)通道,是一種先批準(zhǔn)后驗(yàn)證的藥物監(jiān)管模式。附條件批準(zhǔn)通道適用于治療嚴(yán)重危及生命且尚無(wú)有效治療手段的疾病及罕見(jiàn)病的藥品、公共衛(wèi)生方面急需的藥品。現(xiàn)有臨床研究資料尚未滿(mǎn)足常規(guī)上市注冊(cè)的全部要求,但臨床試驗(yàn)已有數(shù)據(jù)顯示療效并能預(yù)測(cè)其臨床價(jià)值的藥品,可在得到最終的III期臨床結(jié)果之前申報(bào)上市。
圖表6. 納入附條件批準(zhǔn)通道的基本條件
來(lái)源:國(guó)家藥監(jiān)局,中康產(chǎn)業(yè)資本研究中心
附條件批準(zhǔn)通道加快了一批具有顯著臨床價(jià)值創(chuàng)新藥的上市進(jìn)程,一方面藥企可以提早回收研發(fā)成本,改善藥企研發(fā)的可持續(xù)性;另一方面提升了國(guó)內(nèi)患者對(duì)于一些治療嚴(yán)重危及生命的藥品的可及性。尤其是在發(fā)生嚴(yán)重危害公共衛(wèi)生健康的重大疾病時(shí),附條件批準(zhǔn)通道可以加速繼續(xù)藥物或疫苗的上市。近幾年,跨國(guó)藥企和本土藥企都有一些創(chuàng)新藥物、疫苗獲得了附條件批準(zhǔn)上市。
圖表7. 近年部分通過(guò)附條件批準(zhǔn)通道上市的藥物和疫苗
來(lái)源:國(guó)家藥監(jiān)局、公司網(wǎng)站、中康產(chǎn)業(yè)資本研究中心
結(jié)語(yǔ)
2020版《藥品注冊(cè)管理辦法》、化藥和生物制品新分類(lèi)、藥品專(zhuān)利糾紛早期解決機(jī)制,以及審評(píng)特殊通道(優(yōu)先審評(píng)審批、突破性治療藥物、附條件批準(zhǔn)上市)等藥監(jiān)政策,反映出改革方向的轉(zhuǎn)變,已經(jīng)從解決“量”的問(wèn)題轉(zhuǎn)移到提升“質(zhì)”的問(wèn)題,政策更加支持創(chuàng)新程度高、臨床優(yōu)勢(shì)突出、臨床價(jià)值顯著的藥品。藥監(jiān)政策配合醫(yī)保和醫(yī)療政策,將有助于改善臨床用藥結(jié)構(gòu),推動(dòng)價(jià)值醫(yī)療的實(shí)現(xiàn)。
- 下一篇:暫無(wú)
- 上一篇:42種常用藥轉(zhuǎn)換為非處方藥 去藥店不用處方


