8月25日訊 即將于9月1日起執(zhí)行的《基本醫(yī)療保險用藥管理暫行辦法》中提到,《基本醫(yī)療保險藥品目錄》實行通用名管理,該目錄內藥品的同通用名藥品自動屬于基本醫(yī)療保險基金支付范圍。這也意味著在省級帶量采購試點中,“同通用名”背景下中藥和生物制品相互取代。
無獨有偶,8月14日,藥品審評中心發(fā)布關于公開征求《生物類似藥相似性評價和適應癥外推技術指導原則(征求意見稿)》意見的通知,由此藥品的可互換性再一次被熱議。這對于中藥和生物類似藥的立項有何影響?
“可轉換”與“可替代”
[問題] 集中采購屬于“非醫(yī)療轉換”,需要什么條件?
在化學藥領域,專利過期原研藥及仿制藥屬于化學結構穩(wěn)定且單一的藥品。但是,中藥是多組分、多成分組成的藥品,生物類似藥屬于大分子,三者是不同類型的藥品。那么,中藥和生物類似藥真的能像化學仿制藥取代專利過期原研藥那樣具備可互換性?
實現藥品可互換,通常有“可轉換”(switching)和“可替代”(substitution)兩種形式。
“可轉換”指的是處方醫(yī)生決定用某種具有相同治療目的的一種藥物替換另一種藥物。
“可轉換”又涉及“醫(yī)療轉換”和“非醫(yī)療轉換”。“非醫(yī)療轉換”通常是指由于集中采購等付款人綁定政策或臨床供應可用性等客觀原因,對臨床醫(yī)生的處方構成管理限制從而發(fā)生的轉換。集中采購就屬于“非醫(yī)療轉換”。
“可替代”是指在不咨詢處方醫(yī)生的情況下,在藥房層面分配一種藥物而不是另一種等效藥品“可互換”的可能性。
“自動替代”在化學仿制藥是常見的,但中藥之間、生物類似藥和原研生物制品之間只能做到“高度相似”。
《生物類似藥相似性評價和適應癥外推技術指導原則(征求意見稿)》更是定義了“相似性”——生物類似藥與參照藥之間高度相似,在純度、安全性及有效性上不存在有臨床意義的差別。
中藥“可互換”有障礙
[問題] 如何證明具有“可互換”的有效性和安全性?
同通用名中藥的不同廠家生產對應的標準未必一樣。同一通用名的產品都可以申報國家標準的部頒標準,但不同生產廠家的工藝生產、檢測方法特別是有效成分的定量分析未必相同,這意味著起臨床作用的關鍵有效成分未必一樣。整體而言,標準相對較統一的產品是進入中國藥典的中成藥,其處方、制法、性狀和檢測方法基本一致,但即使進入藥典,其標準不一定能完全定量。
以進入2015年版增補本的血塞通片、顆粒、膠囊為例。2019年發(fā)布的《關于血塞通膠囊、血塞通顆粒、血塞通片國家藥品標準修訂草案的公示(第4次)》中提到的血塞通膠囊標準,制法為“取三七總皂苷,加適量輔料混勻或制成顆粒,裝入膠囊”。輔料適量意味著不同廠家可能有不同的輔料,就算輔料體系的成分類似,也會面臨輔料用量不同的問題。最終結果是,不同廠家的藥學部分可能存在差異。而化學藥仿制藥的要求是和專利過期原研藥的輔料體系和輔料用量基本一致。
含量測定標準方面,2020年版藥典對三七總皂苷和血塞通膠囊的要求如表1。而若以“三七總皂苷”這種相對成分比較確定的中藥有效部位作為通用名,首先會遇到一種情況:總皂苷是皂苷類化合物的總稱,往往含有多個不同的成分,鑒于成分太多,指紋圖譜往往選擇最有代表性且有一定藥理作用的成分作為質量控制的檢測物質,這意味著沒有列入質量指標的成分組合很大程度是不同的。
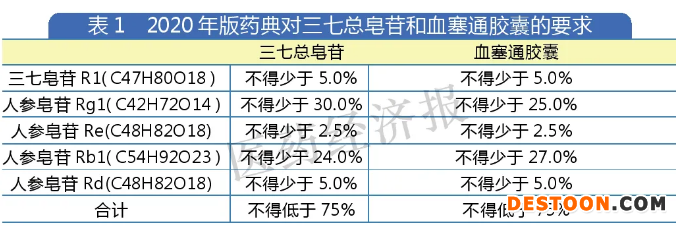
值得注意的是,藥典往往代表的是最低標準,往往是不低于一個百分比值,不同廠家可能在單一成分中各有高低,那么如何比較療效呢?是否含量更高的產品對人體就更好?
此外,三七總皂苷的有效成分檢測要求人參皂苷Rb1不少于24.0%,血塞通膠囊的人參皂苷Rb1不少于27.0%,高于提取物的標準要求。由此可見,一些中藥提取物的關鍵成分可能低于制劑的關鍵成分,制劑在做質量控制時,往往需要提高相應成分質量才能在實際生產上達標。
上述都是關鍵因素,決定中成藥之間是否具有“可互換”的有效性和安全性,因而都需要臨床數據來證實。
生物類似藥需要證明“可互換”
[問題] 臨床試驗高成本+集采斷崖式降價,何解?
按《生物類似藥相似性評價和適應癥外推技術指導原則(征求意見稿)》,生物類似藥相似性評價應基于藥學、非臨床、臨床比對研究結果進行綜合評價,以確定整體相似性。
前期研究結果顯示,候選藥與參照藥之間存在差異,但該差異對安全有效性的影響尚不確定的,在經后續(xù)針對性對比研究后,未發(fā)現其具有臨床意義的影響,如二者微小的質量差異未發(fā)現對安全性、有效性和免疫原性等存在影響的,則可認為具有相似性。
如存在質量差異,且該差異對安全性、有效性和免疫原性等影響存在不確定性的;或體外生物學活性評價模型對體內行為和藥效預測有限的,則應有針對性地開展擴展的藥學比對試驗,以及必要的非臨床/臨床比對試驗,以評估并證明該質量差異對有效性和安全性的作用,不影響整體相似性。
如質量差異對臨床安全性和有效性產生影響,則不宜按生物類似藥進行研發(fā)。
由此可見,臨床試驗的研究結果才是決定是否“相似”的關鍵。
臨床相似性評價包括PK相似性評價、PD和(或)PK/PD相似性評價、有效性相似性評價、安全性相似性評價和免疫原性相似性評價。這些評價可以在上市前一并研究。而在我國,只要上市獲批了,就有可能面臨進入國家級或省級的集中采購,藥品價格面臨腰斬。
結語
綜上所述,若中藥和生物制品實施集中采購的模式,依然“唯低價是取”,那么企業(yè)只要滿足藥典最低指標進行成本控制,而這未必是中藥和生物制品價格管理的最佳模式。
特別是對于一些已經完成中藥大品種研究、從質量成分的指紋圖譜到臨床試驗數據都建立一套完整標準的中藥,更加不利。而且,這給行業(yè)一個不好的信號——完成全面的科學的質量和療效分析也要面臨價格競爭的壓力,最終醫(yī)保愿意買單的卻是最低價但研究未透徹的中藥。


 110102000668(1)號
110102000668(1)號